RedAcu.Com
Promoting Self-Healing
[ Chaperones Policy ]

PATIENT RIGHTS – USE OF CHAPERONES DURING MEDICAL EXAMINATIONS/ TREATMENTS
The AMA (American Medical Association) code of ethics states that physicians should adopt a policy whereby patients have the right to request a chaperone during physical examinations/ treatments. Some states have adopted a legal policy for physicians to provide a chaperone for intimate examinations/ treatments.
The Texas State Medical Board states that:
“A policy that patients are free to make a request for a chaperone should be established in each health care setting. This policy should be communicated to patients, either by means of a well-displayed notice or preferably through a conversation initiated by the intake nurse or the physician. The request by a patient to have a chaperone should be honored.”
An opportunity for private conversation with the patient should be provided without the chaperone present, and physicians should minimize history taking and sensitive inquiries while the chaperone is present for the exam. Your chaperone should be your friends(trained health professionals) or family members.
You have a right to refuse a chaperone as well.
Please feel free to change your decision anytime.
[ Medical records or Payment records must be picked up in-person by patients only.]
These policies are to assure both the patient and Dr. Kim that communications are professional and respectful. We encourage patients to know their rights, and to share this information with family and friends.
[ Cancellation/Change/Missed Appointment Policy ]
Our goal is to provide quality medical care in a timely manner. In order to do so, we have had to implement an appointment/cancellation/change policy. This policy enables us to better utilize available appointments for our patients in severe pain needing immediate care.
Cancellation/Change of an Appointment: In order to be respectful of the medical needs of other patients, please be courteous and call the office promptly if you are unable to attend an appointment. This time will be reallocated to someone who is in urgent need of treatment. If it is necessary to cancel/change your scheduled appointment, we require that you call or text at least 48 hours in advance. Calling or texting early in the day is appreciated. Your early cancellation/change will give another person the possibility to have access to timely medical care.
" There is nothing to compare with your health. "
25222 Grogan's Mill Road, The Woodlands, Texas 77380
www.RedAcu.com
[ Patient Non-Discrimination Policy ]
The Woodlands Dr.Kim's Acupuncture Health Center is committed to equal care for all Dr. Kim's patients.
Dr. Kim's Patient Non-Discrimination Policy was established to protect the well-being of every patient under Dr. Kim's care.
The patient has the right to treatment without discrimination as to race, age, religion, sex, national origin, socioeconomic status, culture/creed, education, sexual orientation, gender identity or expression, disability, veteran status, or source of payment. You will be treated with dignity, compassion, and respect as an individual.
[ Dr. Kim's Acupuncture Treatment Policy ]
Before Treatment
1. Wear loose, clean, and comfortable clothing for easy access to acupuncture points.
2. Bring a medical diagnosis you have gotten from a Doctor (M.D., D.O., DC., or DDS), if you can.
3. Bring Blood tests, MRI scan (CD), CT scan, EKG(ECG), X-ray results, Ultrasound, ..., if you have.
4. Don't eat large meals just before or after your visit.
5. Each Acupuncture Treatment takes 45min to 1hour. Sometimes more than 1hour.
6. Your first visit will be Dr.Kim's Acupuncture Treatment,Medical Exam,Tongue and Pulse Diagnosis,Health History Review,Thermography scans/analysis, Consultation with your condition,Treatment Plan with your condition, and Wellness Check (Diet, Lifestyle, ...),
Plus, Neuro-Ear Therapy if you need.
7. Your follow-up visit will be Medical Exam/Wellness Check and Dr. Kim's Acupuncture Treatment.
(Thermography or Neuro-Ear Therapy depends on patient's condition)
8. Dr. Kim doesn't accept any insurance.
9. Read the payment-price carefully before booking an appointment.
After Treatment
1. Keep Thermography (inflammation) scan images in your cell phone. Dr. Kim doesn't keep yours.
2. Drink more warm water as much as you can.
3. Refrain from overexertion, working out, sexual intercourse, or alcohol for up to 12 hours after visit.
4. Avoid stressful situations, Make time to relax, and be sure to get plenty of rest.
5. Between visits, take notes of any changes that may have occurred, such as the alleviation of pain,pain moving to other areas,small bruises(Ecchymosis),big bruises,swollen tissues or changes in the frequency and type of problems.
6. Do your best to understand the inflammation that you have in your body !!!
You will save lots of money and time !!!
7. Maximize the quality of sleep.
8. Medical records or Payment records must be picked up in-person by patients only.
Please read the policies completely before booking an appointment
APPOINTMENT : 832) 660 - 4838 TEXT OR CALL
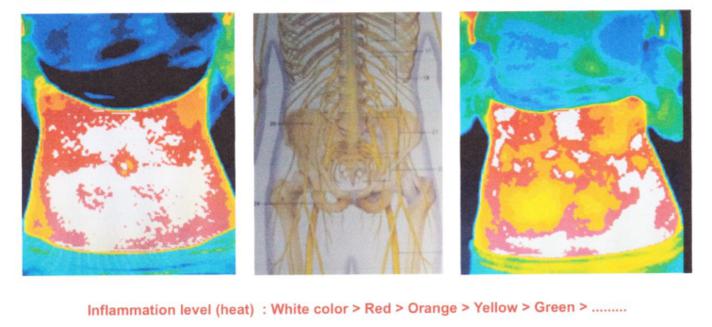
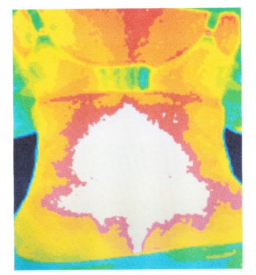

Thermography Scan / Study
Thermal imaging is used to study a broad number of diseases where skin temperature can reflect the presence of inflammation in underlying tissues or where blood flow is increased or decreased due to a clinical irregularity.
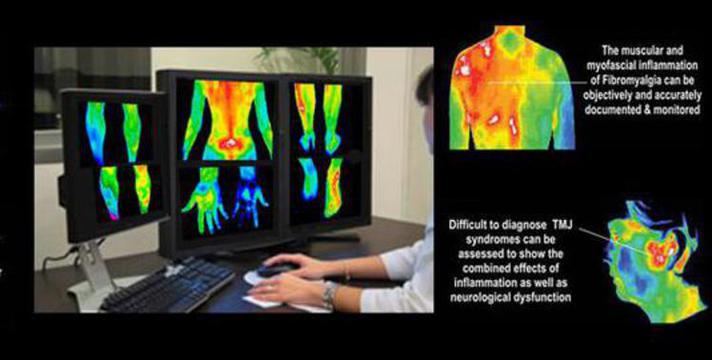
Thermography is a non-invasive, safe, diagnostic tool that takes pictures and analyzes changes in the skin’s surface temperature without the use of radiation. While X-rays, ultrasound, and mammography show us the structure of the body, they will miss such things as active inflammation and increased blood supply as found in many illnesses. Thermography is a unique technology that takes a picture and creates a map of the infrared patterns of the body. It is different from other screening tools because it helps you to see how your body functions.